News
Feb 2025, Haoran, Dingjie, Qi, Puwen, Yunhao, Xiaoyu's study of miniQuant and K-value for gene isoform quantification has been accepted in principle by Nature Biotechnology! Congratulations!
 Try miniQuant
Try miniQuant
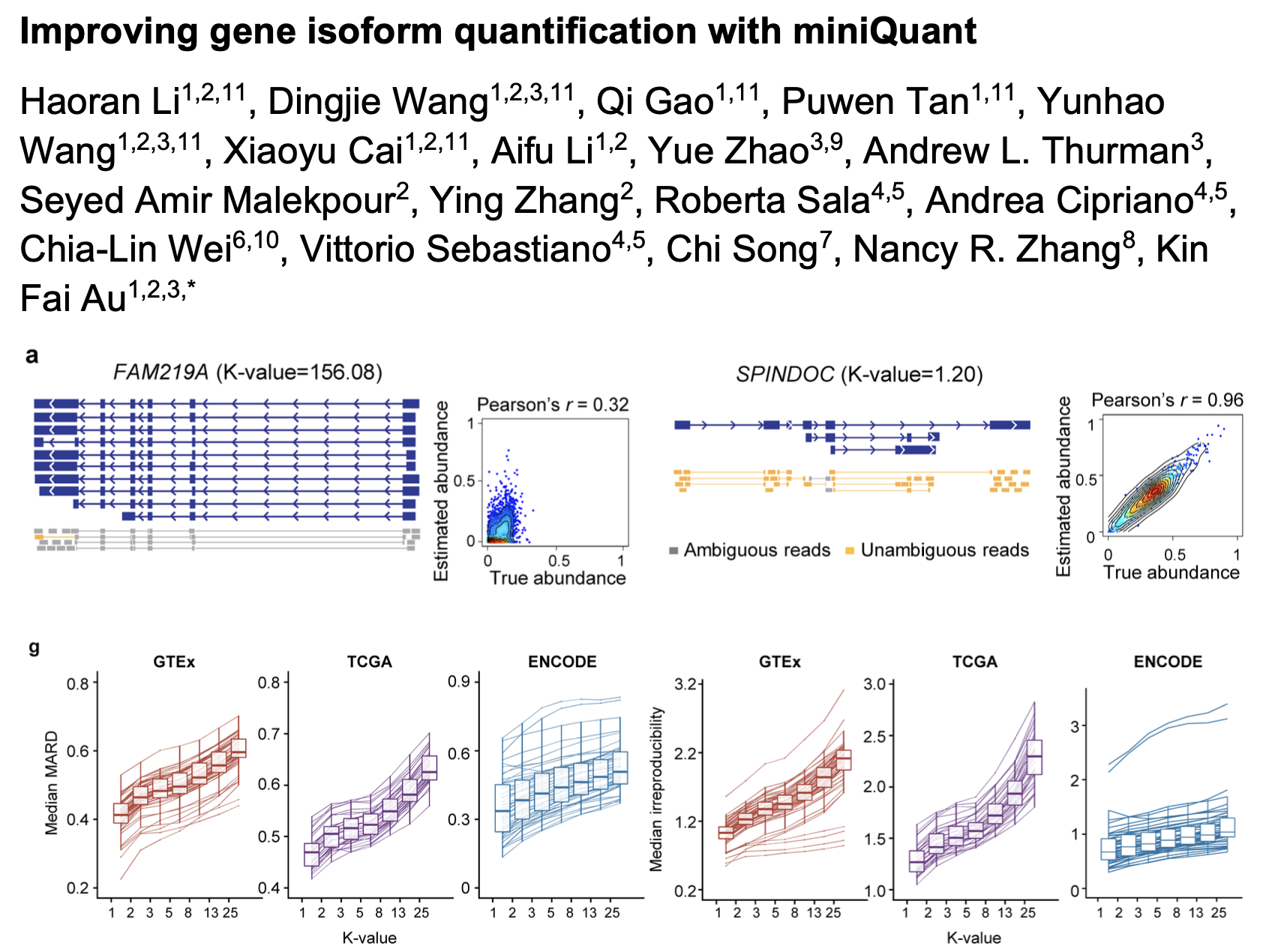
Feb 2025, Bo, Dingjie and our collaborators’ paper of zygotic transposable elements activation during zebrafish early embryogenesis has been accepted in principle by Nature Communications! Congratulations!
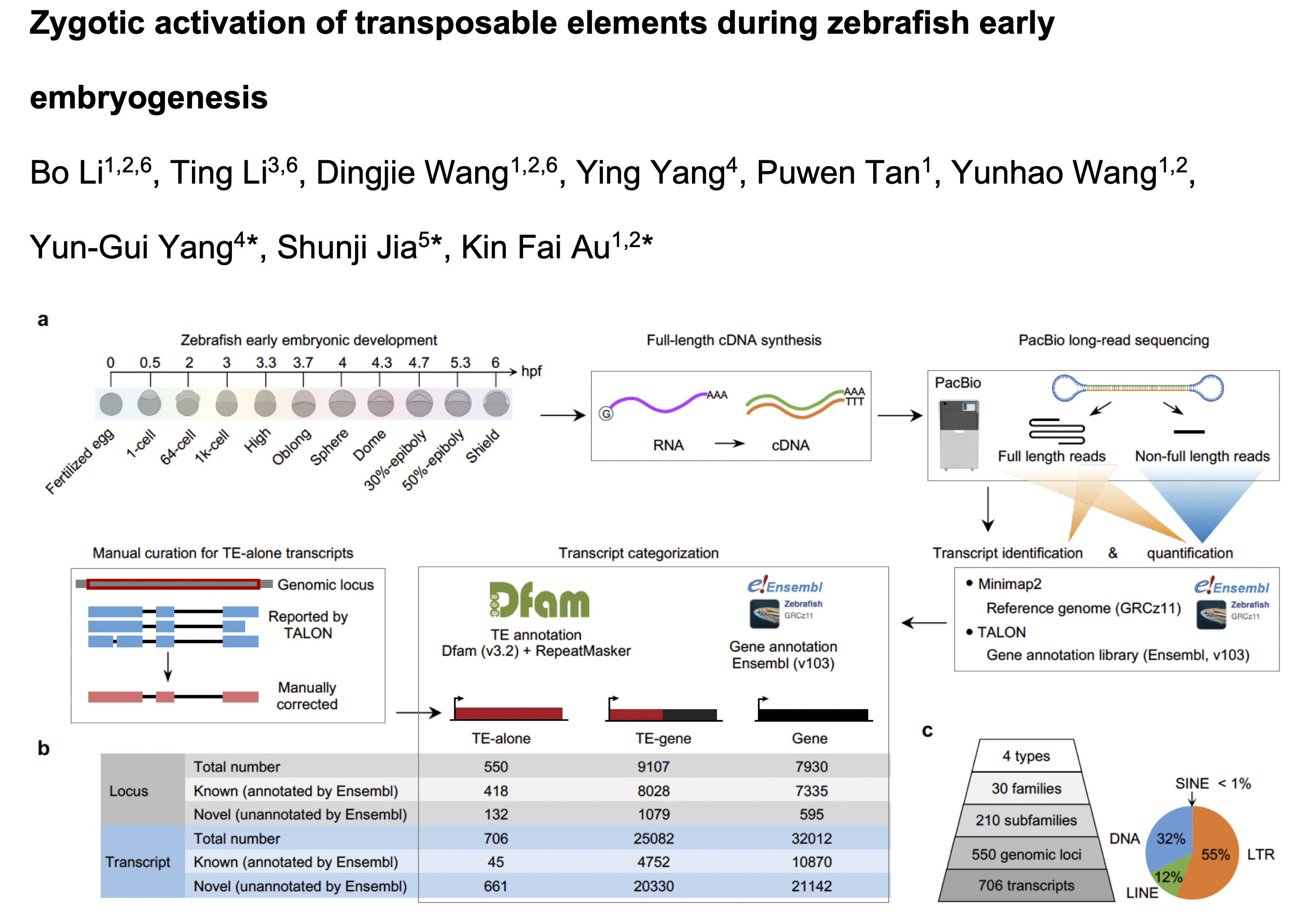
April 2023, Yunhao and our collaborators’ single-cell RNA m6A mapping study has been accepted in principle by Nature Biotechnology! Congratulations!

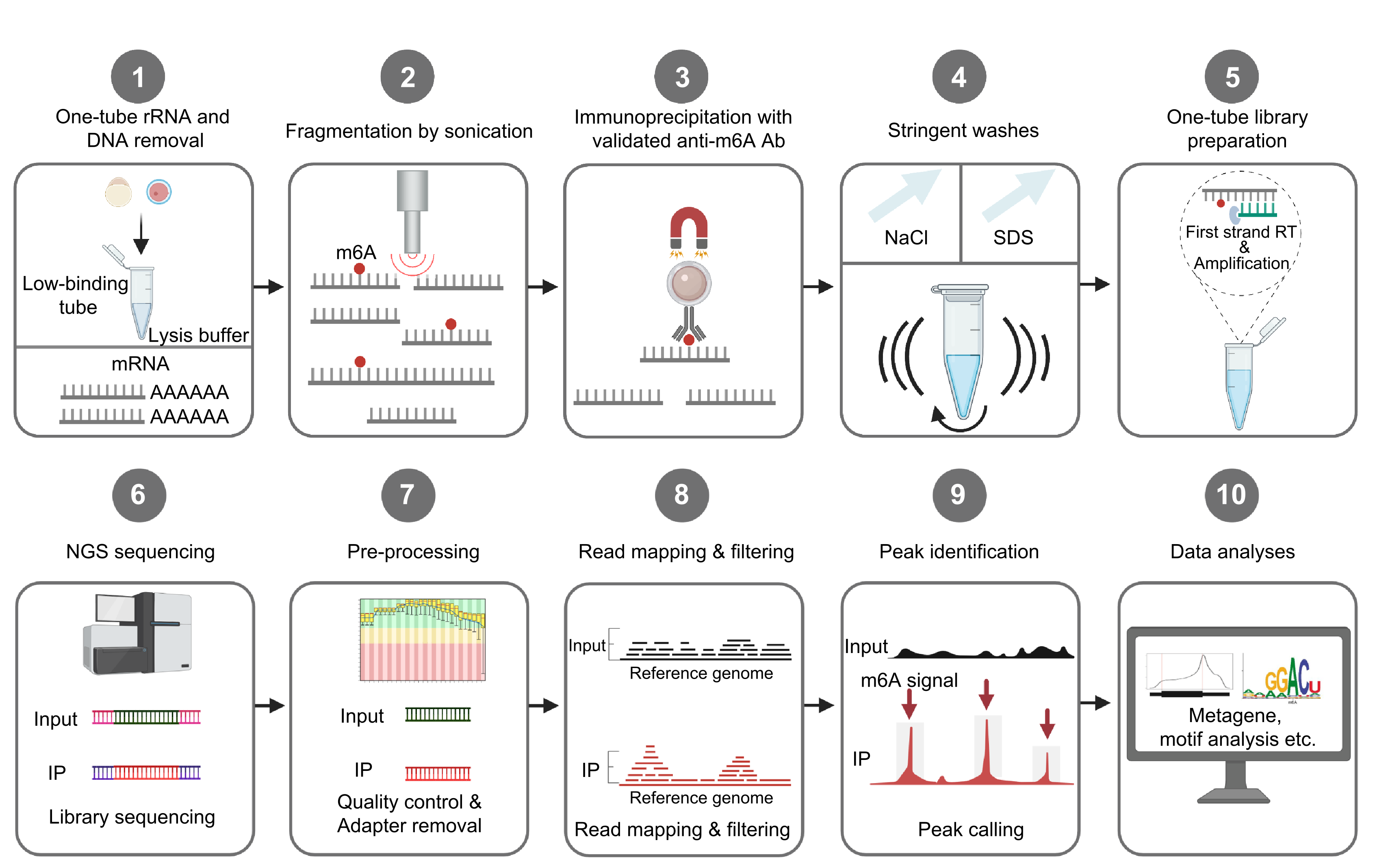
Feb 2023: Yunhao and our collaborators’ RNA m6A work was accepted in principle by Nature Structural & Molecular Biology! Congratulations!

Feb 2023: We are thrilled to join the Department of Computational Medicine and Bioinformatics at University of Michigan and start a new journey of more exciting researches!
We are also recruting talented students and postdoctoral scholars to join our team. For more information, please refer to our homepage and feel free to contact us!
July 2022: Kin Fai is appointed as the Vice Chair of Research at the Department of Biomedical Informatics. Congratulations!
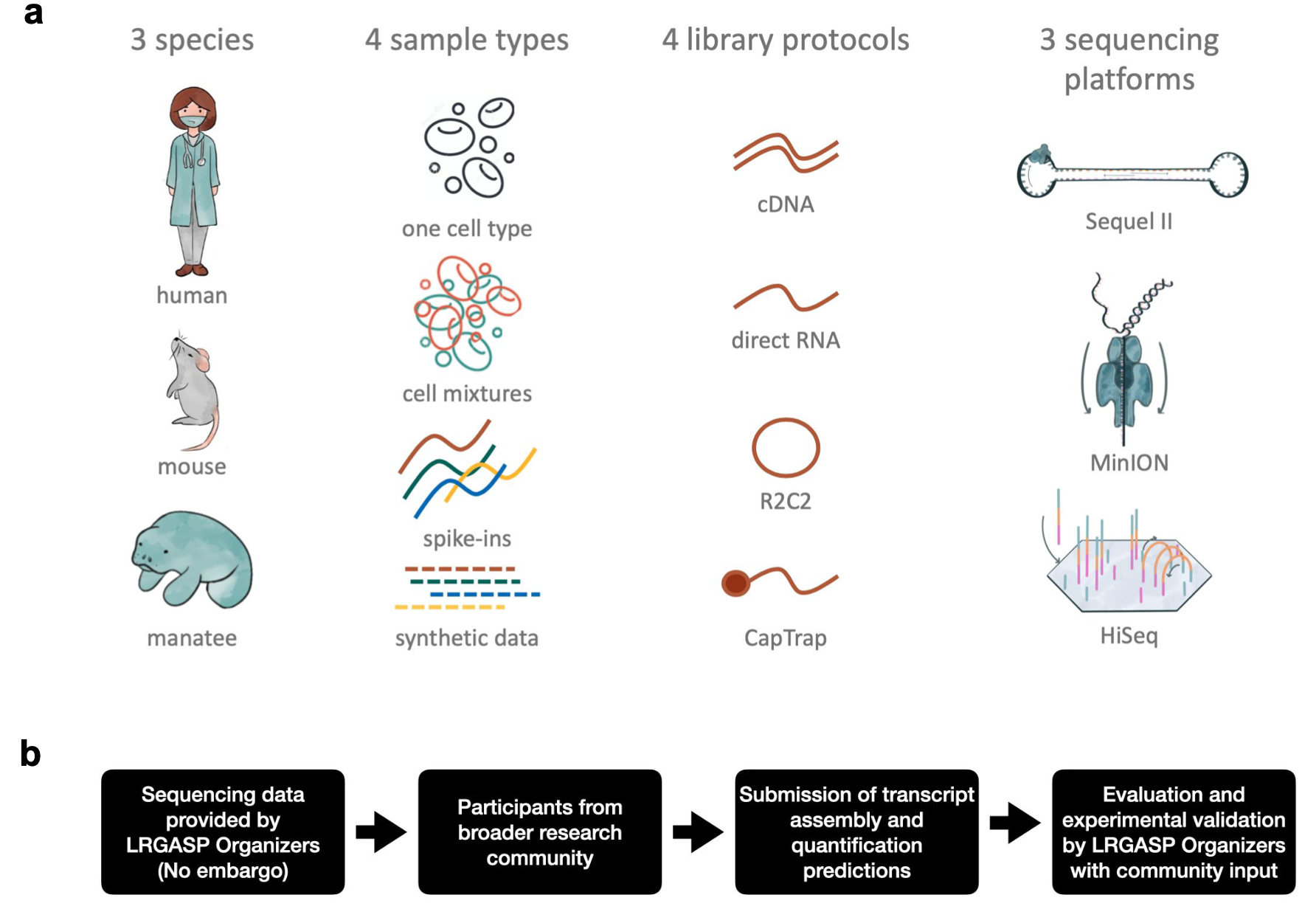
May 2022: Dingjie and Haoran's latest manuscript of LRGASP project was accepted in principle by Nature Methods! Congratulations!
Oct. 12th, 2021: The Au Lab receives two NIH R01 grants from National Human Genome Research Institute and National Institute of General Medical Sciences to develop a series of bioinformatics tools to study gene isoforms and transposable elements in human stem cells and germ cells.
Jul. 15th, 2021: The Au Lab receives an NIH R01 grant from NATIONAL HUMAN GENOME RESEARCH INSTITUTE to develop a series of bioinformatics tools to analyze gene isoforms in human stem cells
Jul. 12th, 2021: Congratulation on Haoran's paper accepted by Bioinformatics!
Zhang, H.*, Li, H.*,Jain, C., Cheng, H., Au, K.F.**, Li, H.**, Aluru, S.**
Bioinformatics. 2021. [Manuscript]
* These authors contributed equally to this work.
** Co-corresponding author
Jul. 1st, 2021: Kin Fai serves a regular member at the NIH Genomics, Computational Biology and Technology Study Section.
Dec. 16th, 2020: Congrats! Kin Fai receives the OSU-BMI Award for Research Excellence!
Dec. 15th, 2020: Congratulations to Yunhao, Audrey, Diana and Yuru for their article accepted by Nature Biotechnology!
Wang, Y., Zhao, Y., Bollas, A., Au, K.F.#
in press, Nature Biotechnology. 2020
# Corresponding author
Sun, Y.H.*, Wang, A.*,Song, C., Srivastava, R.K., Au, K.F.**, Li, X.Z.**
Nature Communications. 2020. [Manuscript]
* These authors contributed equally to this work.
** Co-corresponding author
Dec. 16th, 2020: Congrats! Kin Fai receives the OSU-BMI Award for Research Excellence!
Dec. 15th, 2020: Congratulations to Yunhao, Audrey, Diana and Yuru for their article accepted by Nature Biotechnology!
Wang, Y., Zhao, Y., Bollas, A., Au, K.F.#
in press, Nature Biotechnology. 2020
# Corresponding author
Jun 10th, 2020: New Publications.
Wang, D., Zou, X., Au, K.F.#
Methods. 2020. [Manuscript]
# Corresponding author
March, 2020: Congrats! Professor Au joined the editorial boards of the premier journals of genomics and bioinformatics.
 Kinfai joined the editorial board of Genome Biology .
Kinfai joined the editorial board of Genome Biology .
 Kinfai joined the editorial board of Genome Research.
Kinfai joined the editorial board of Genome Research.Jan 17th, 2020: New Publications.
Wang, A., Au, K.F.#
Genome Biology. 2020. [Manuscript]
# Corresponding author
Audrey presents our recent work at the Nanopore Community Meeting 2019.

Jun 14th, 2019: Our latest paper of identifying nucleosome occupancy and chromatin accessibility on single DNA molecule is selected as the cover of the August Issue of Genome Research.
Single-molecule long-read sequencing reveals the chromatin basis of gene expression.
Wang, Y., Wang, A., Liu, Z., Thurman, A., Powers, L.S., Zou, M., Hefel, A., Li, Y., Zabner, J., Au, K.F.
Genome Res. Cover photo of issue August 2019 . [Manuscript]
Jun 15th, 2018: Shuhua Fu, Anqi Wang and Andrew Thurman are going to make presentations in ISMB 2018, July 7th, Chicago.
IDP-denovo: de novo transcriptome assembly and isoform annotation by hybrid sequencing. (Shuhua Fu)
Theoretical analysis of graph-based and alignment-based hybrid error correction methods for error-prone long reads. (Anqi Wang)
Gene isoform abundance quantification with Third Generation transcriptome sequencing. (Andrew Thurman)
Feb 23rd, 2018: IDP-denovo paper is published.
Fu, S., Ma Y., Yao, H., Xu, Z., Chen, S., Song, J., Au, K.F.
Bioinformatics 2018. [Manuscript]