News
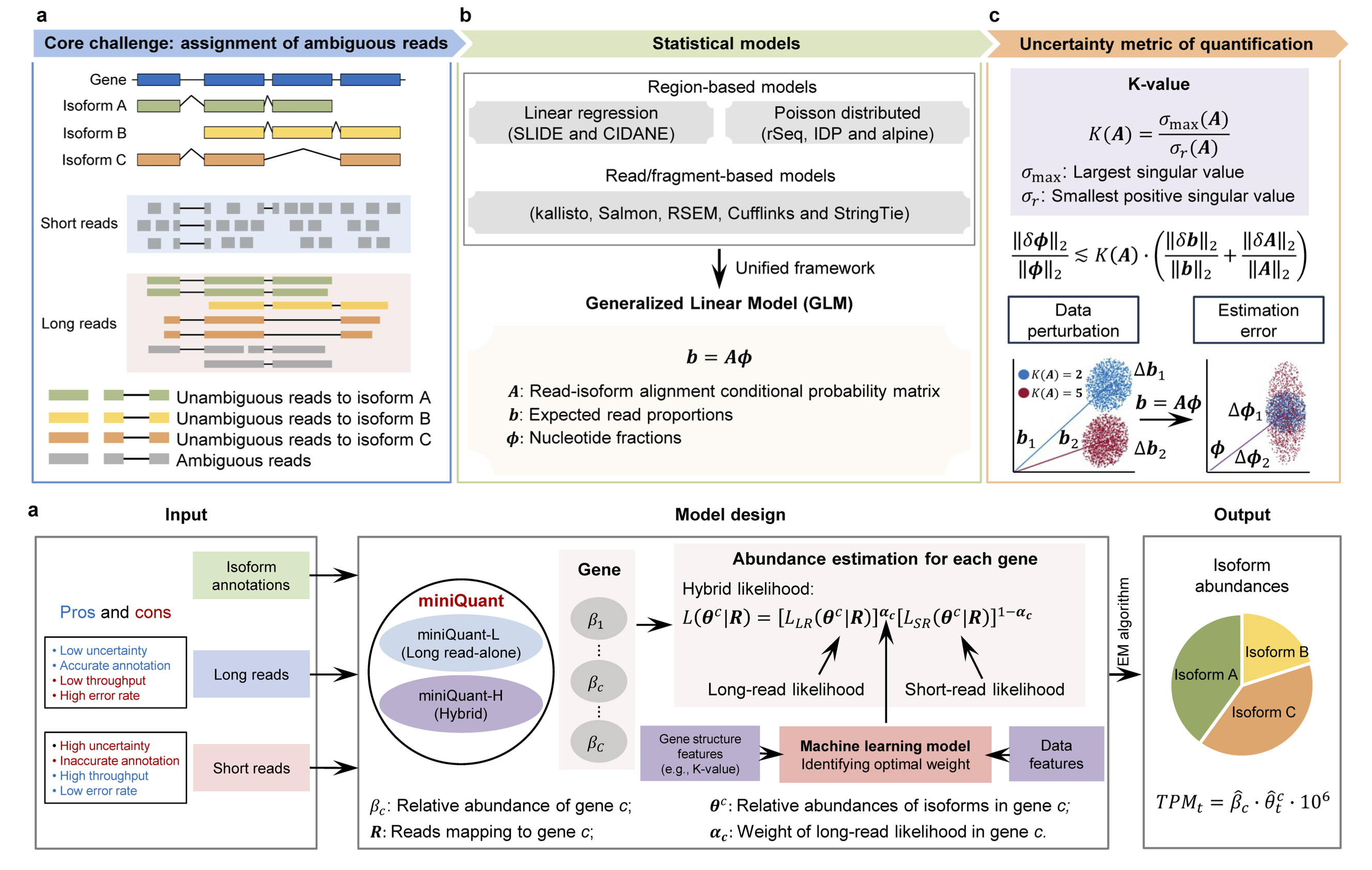
Feb 2025, our paper of miniQuant and K-value for gene isoform quantification has been accepted in principle by Nature Biotechnology! Congratulations!
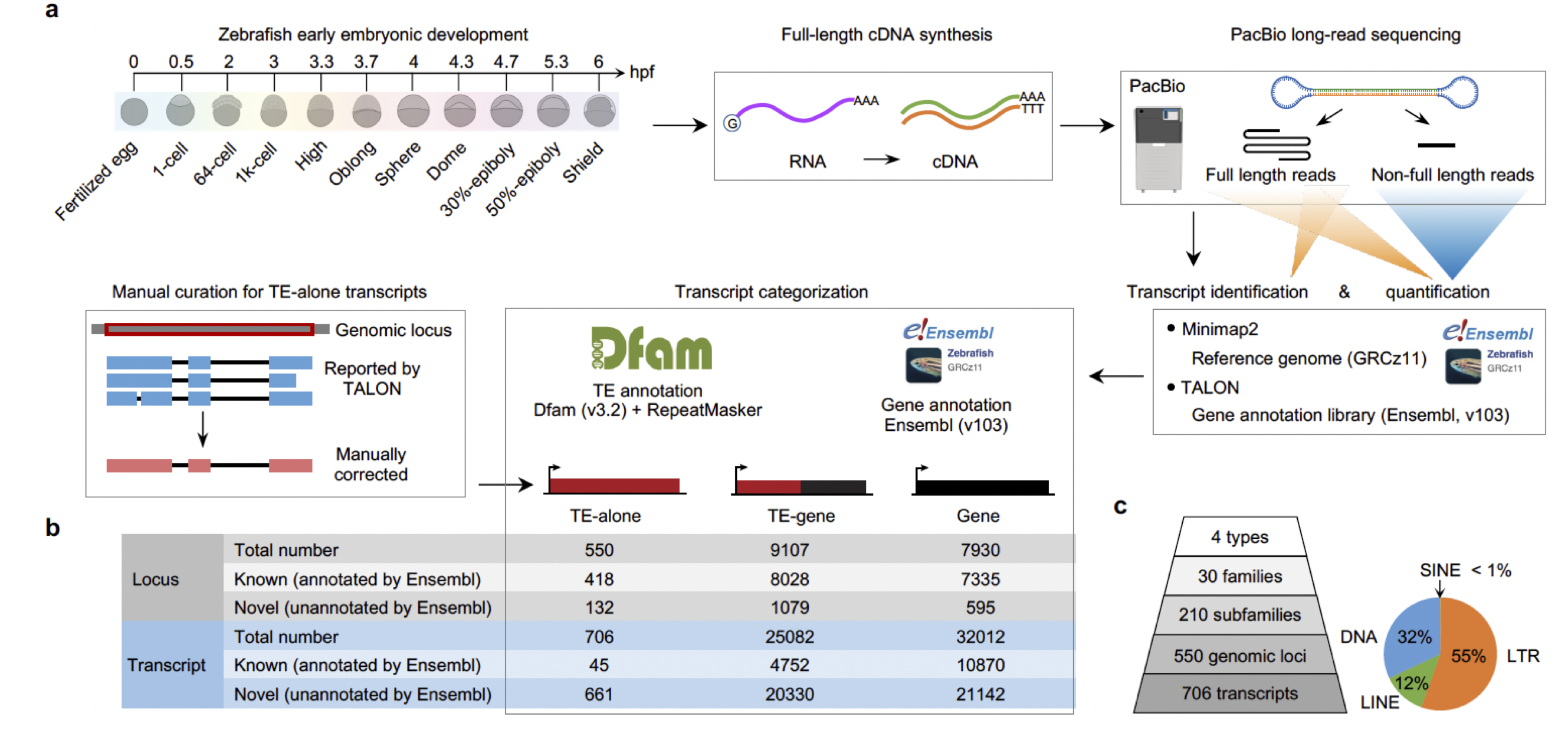
Feb 2025, our paper of zygotic transposable elements activation during zebrafish early embryogenesis has been accepted in principle by Nature Communications! Congratulations!
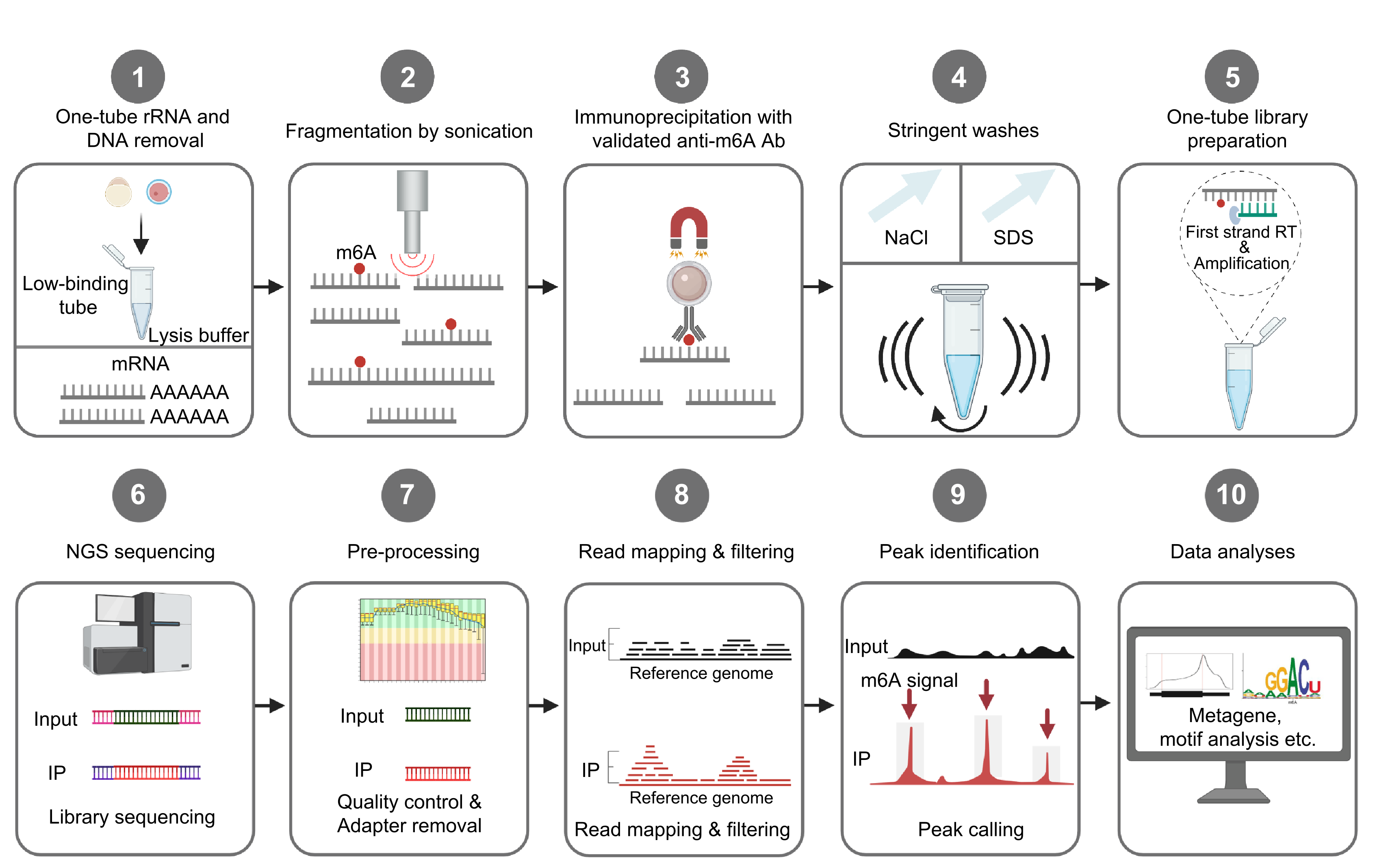
April 2023, Yunhao and our collaborators’ single-cell RNA m6A mapping study has been accepted in principle by Nature Biotechnology! Congratulations!
Recent publications
Zygotic activation of transposable elements during zebrafish early embryogenesis
The RNA m6A landscape of mouse oocytes and preimplantation embryos
Wang, Y.*, Li, Y.*, Skuland, T., Zhou, C., Li, A., Hashim, A., Jermstad, I., Khan, S., Dalen, K. T., Greggains, G. D., Klungland,A.**, Dahl, J.A.**, Au, K.F.**
Nature Structural & Molecular Biology. 2023.[Manuscript]Notice
Kin Fai is organizing a special issuse of long-read sequencing in Genome Biology. We are looking
forward to your exciting work of long reads!
https://genomebiology.biomedcentral.com/Long-Read
Open Positions
Strong ability in statistical analysis or computer programming (python/perl, C/C++, R, Matlab) is desirable.
The postdoctoral research scholars will have the opportunity to collaborate with internationally recognized biomedical scientists at University of Iowa Carver College of Medicine, Stanford University, University of Oxford and Pacific Biosciences.